Что нужно знать о эпилепсии
пациентам и специалистам

Диагностика пароксизмальных состояний в неврологии — одна из самых сложных проблем.
УДК 616.853
DOI: 10.32471/umj.1680-3051.135.171088
Т.А. Литовченко
Харьковская медицинская академия последипломного образования
Особые трудности вызывает дифференциальная диагностика эпилептических и неэпилептических пароксизмальных нарушений, учитывая отсутствие абсолютно достоверных клинических маркеров заболевания в межпароксизмальный период, не всегда достаточную информативность инструментальных методов исследования и недостаточную осведомленность врачей.
Международная противоэпилептическая лига (International League Against Epilepsy — ILAE, 1997) определяет имитаторы эпилепсии (заболевания и состояния, которые могут быть ошибочно диагностированы как эпилепсия) как «заболевания (состояния), клиническая манифестация которых предполагает не связанные с патологической сверхмерной активностью нейронов головного мозга (а) нарушения функций головного мозга (головокружение, синкопе, нарушения сна, двигательные нарушения, транзиторная глобальная амнезия, мигрень, энурез) и (b) псевдосудороги (неэпилептические внезапные эпизоды изменения поведения психогенного происхождения, которые могут сочетаться с эпилептическими приступами).
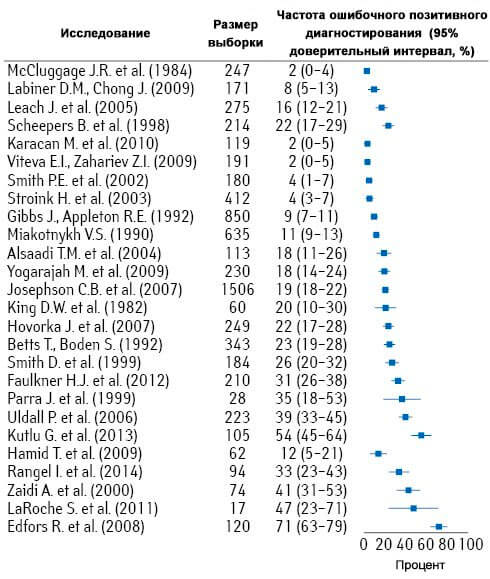
Неэпилептические пароксизмальные нарушения ошибочно диагностируют как эпилепсию в 20–30% случаев даже в медицинских центрах третьего уровня (Gates J.R., 2002; NICE, 2004). По данным последних метаанализов, ложную положительную диагностику эпилепсии отмечают в 2–71% случаев (Xu Y. et al., 2016). Кроме того, не менее 30% пациентов с эпилепсией имеют также неэпилептические (чаще психогенные) приступы (Panayiotopoulos C.P., 2010; Xu Y. et al., 2016). Существует целый ряд нормальных физиологических пароксизмальных феноменов, которые также могут осложнить диагностику (например, содрогание во время сна и т.п.). Y. Xu и соавт. (2016) в своем обзоре приводят данные, свидетельствующие о значительном количестве ошибочного диагностирования эпилепсии даже в специализированных высококвалифицированных медицинских учреждениях (рис. 1).
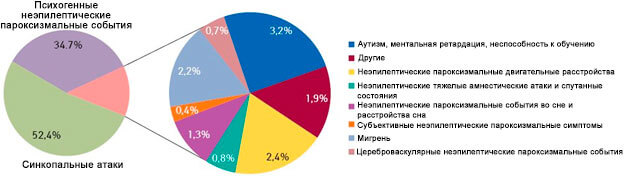
Чаще всего проблему дифференциальной диагностики составляют психогенные неэпилептические припадки и синкопе, но целый ряд двигательных нарушений, имеющих пароксизмальные проявления, могут быть ложно диагностированы как эпилептические приступы, и наоборот (рис. 2).
Двигательные нарушения, характеризующиеся внезапными преходящими эпизодами патологической непроизвольной двигательной активности, такие как хорея, атетоз, неэпилептические миоклонии, дистония, пароксизмальная дискинезия, нарушение координации произвольных двигательных актов или объединенные нарушения координации и непроизвольной двигательной активности (пароксизмальная атаксия) представляют собой значительные диагностические трудности даже для высококвалифицированных специалистов (Panayiotopoulos C.P., 2010; Wolters E., Baumann C. (Eds.), 2014; Xu Y. et al., 2016; Greenland J.C., Barker R.A. (Eds.), 2018). Такие заболевания могут быть наследственными, возникать случайным образом по неопределенным причинам или быть вторичными, вследствие других заболеваний и патологических состояний.
Неэпилептические пароксизмальные двигательные нарушения по любой причине являются довольно частыми имитаторами эпилептических приступов. Также эпилепсия, в ряде случаев, может имитировать некоторые двигательные нарушения. Значительные трудности вызывает дифференциальная диагностика миоклонуса, которая может иметь физиологическое происхождение (например, гипнагогический миоклонус не является патологией), быть проявлением ряда патологических состояний — неэпилептический (субкортикальный) миоклонус и похожие на миоклонии неэпилептические немиоклонические двигательные феномены (Темин П.А., Никанорова М.Ю. (ред.), 1999; Panayiotopoulos C.P., 2010; Greenland J.C., Barker R.A. (Eds.), 2018; ILAE, 2019). Нередко эпилептические миоклонии не фиксируются пациентами и не диагностируются врачами вовремя.
Эпилептические миоклонии могут быть единственным проявлением эпилептического приступа или наблюдаться в структуре других типов приступов. Эпилептический миоклонус чаще имеет первично-генерализованное происхождение и наблюдается при многих идиопатических синдромах генерализованных эпилепсий (ювенильная миоклоническая эпилепсия, эпилепсия с миоклонически-астатическими приступами, доброкачественная миоклоническоая эпилепсия раннего возраста и т.п.). При эпилептическом миоклонусе мышечные сокращения следуют за пик-волновыми изменениями электроэнцефалограммы (ЭЭГ) с интервалом 50 мс. При фокальных эпилепсиях (эпилепсия Кожевникова, синдром Расмусена) эпилептический миоклонус имеет кортикальное происхождение вследствие стимуляции сенсомоторной коры, по механизму он фокальный или мультифокальный. Ретикулярный миоклонус имеет генерализованный механизм и наблюдается при ряде форм генерализованной эпилепсии.
Неэпилептический миоклонус (его также называют миоклоническими гиперкинезами) имеет субкортикальный механизм, мышечные сокращения не сопровождаются характерными изменениями биоэлектрической активности головного мозга (нет пик-волновой активности на ЭЭГ) (Темин П.А., Никанорова М.Ю. (ред.), 1999; Chowdhury F.A. et al., 2008; Dalla Bernardina B., 2009; Panayiotopoulos C.P., 2010; Chitre M., 2013; ILAE, 2019). По механизму может быть подкорковым, стволовым и спинальным:
Дифференциальная диагностика этих патологических и физиологических состояний чрезвычайно сложна, тем более что даже неэпилептические миоклонии нередко дают терапевтический ответ при лечении противоэпилептическими препаратами (ПЭП). Но есть ряд клинических отличий эпилептических и неэпилептических миоклоний:
Помимо миоклоний, существует ряд неэпилептических двигательных нарушений, которые необходимо дифференцировать от эпилепсии. Самые частые из них — тремор, тики, самопроизвольные движения.
Тики — короткие, внезапные, непроизвольные, неритмичные, повторяющиеся и стереотипные движения или звуки, которые могут появляться периодически или постоянно. Тики могут быть простыми или сложными. Простые тики привлекают только одну группу мышц, они короткие и бессмысленные. Сложные тики могут длиться дольше и оказываться более целеустремленными (стереотипная последовательность движений, слова, целые фразы). Такие расстройства обычно начинаются в детском или юношеском возрасте, усиливаются при стрессе, эмоциях. Моторные проявления при тиках продолжаются >200 мс, с частотой, которая может быть изменена произвольно (Темин П.А., Никанорова М.Ю. (ред.), 1999; Panayiotopoulos C.P., 2010; Wolters E., Baumann C. (Eds.), 2014; ILAE, 2019). Тики обычно появляются в возрасте 5-7 лет, с пиком развития в 9-11 лет. В 50–70% случаев тики сначала двигательные, с преобладанием на уровне лица, плеч или шеи.
Диагноз тиков вполне клинический, и во многом основан на жалобах пациентов: возможность частичного контролирования движений или вокализации, обычно предчувствие развития, чувство облегчения после окончания эпизода.
Для тиков, в отличие от эпилепсии, характерно вовлечение нескольких групп мышц (например, сочетание моргания с наморщиванием лба, носа, пожимание плечами и т.п.). Во время тика пациент не прерывает обычную двигательную активность и разговор, может по заданию врача воспроизвести гиперкинез, что невозможно для пациентов с эпилепсией. При коротких стереотипных гиперкинезах по типу моргания или прикрывания глаз с заведением глазных яблок для уточнения диагноза необходимо проведение ЭЭГ-видеомониторинга (одновременное появление «гиперкинеза» и генерализованной пик-волновой активности свидетельствует о наличии абсанса). Также необходимо проведение ЭЭГ с провокационными пробами.
Нельзя назначать лечение ПЭП ex juvantibus до установления диагноза, поскольку большинство ПЭП также эффективны при тиках, а бензодиазепины, применяемые для лечения пациентов с тиками, могут снижать частоту абсансов (Темин П.А., Никанорова М.Ю.) (ред.), 1999; Panayiotopoulos CP, 2010; Roessner V. et al., 2011; Pringsheim T. et al., 2012).
Основные дифференциально-диагностические клинические и нейрофизиологические признаки эпилепсии и тиков представлены в табл. 1.
| Показатель | Эпилепсия | Тики |
|---|---|---|
| Стереотипность | Да | Да |
| Длительность | 1–3 мин/абсансы до 30 с | Короткие, разной продолжительности, от секунд до нескольких минут |
| Частота | От нескольких в месяц до десятков на сутки | Частые |
| Семейный анамнез | Часто – семейная история эпилепсии | Нет (возможно, очень редко) |
| Возраст дебюта | Часто – в детстве и подростковом возрасте | Чаще 5–12 лет |
| Нарушение сознания | Часто | Никогда |
| Амнезия | Часто | Никогда |
| Провоцирующие факторы | Отсутствуют (в некоторых случаях – фотостимуляция, гипервентиляция) | Стресс, эмоции |
| Электромиография | Короткие вспышки длительностью <50 мс, иногда – до 100 мс | >200 мс, частота может меняться произвольно |
| Возможность произвольного контроля и воспроизведения | Нет | Да |
| Прерывание обычной моторной и речевой активности | Да | Нет |
| Привлечение групп мышц | Только одна группа | Как правило, несколько |
| ЭЭГ | Эпилептиформные феномены (спайки, полиспайки, спайк-волновые комплексы), после приступа – супрессия активности | Без изменений |
Проблему дифференциальной диагностики эпилепсии также может составлять семейный пароксизмальный дистонический хореоатетоз (пароксизмальная некинезиогенная дискинезия, пароксизмальная некинезиогенная дистония, синдром Маунта — Ребека) — неэпилептическое гиперкинетическое расстройство движения, связанное с хромосомой 2q35. Заболевание характеризуется эпизодами непроизвольных хореиформных и дистонических движений, баллизма с началом в детском возрасте. Двигательные приступы достаточно длительные – от ½ ч до нескольких часов, при этом между приступами у ребенка не возникает никакой двигательной или иной патологии. Частота приступов – до нескольких раз в неделю. Нарушения сознания во время приступов не наблюдается, ЭЭГ во время приступа и после него остается неизменным (Chatterjee A. et al., 2002; Ohmori I. et al., 2002; Tsai J.D. et al., 2005; Kato N. et al., 2006; Panayiotopoulos CP, 2010; Wolters E., Baumann C. (Eds.), 2014). Приступы провоцируются разными факторами (кофеин, алкоголь, эмоции). В отличие от эпилептических приступов (при лобной эпилепсии), такие приступы почти у всех пациентов облегчаются или исчезают после короткого периода сна (табл. 2).
| Показатель | Эпилепсия | Семейный пароксизмальный дистонический хореоатетоз |
|---|---|---|
| Стереотипность | Да | Да |
| Длительность | Как правило, 1–3 мин/абсансы до 30 с | От ½ ч до нескольких часов |
| Частота | Разная — от нескольких на год до десятков в сутки | Несколько в неделю |
| Семейный анамнез | Часто | Да (связан с хромосомой 2q35) |
| Возраст дебюта | Часто в детстве и подростковом возрасте | Обычно 5–12 лет |
| Нарушения сознания | Часто | Никогда |
| Амнезия | Часто | Никогда |
| Провоцирующие факторы | Отсутствуют (в некоторых случаях – фотостимуляция, гипервентиляция) | Разные (кофеин, алкоголь, стресс, эмоции) |
| ЭЭГ | Эпилептиформные феномены (спайки, полиспайки, спайк-волновые комплексы), после приступа – супрессия активности | Без изменений |
| Связь со сном | Нередко при пробуждении (лобные припадки) | Облегчаются или исчезают после короткого периода сна |
Пароксизмальная кинезиогенная дискинезия (неэпилептический пароксизмальный кинезиогенный хореоатетоз) характеризуется периодическими короткими эпизодами самопроизвольных движений, вызванных внезапным произвольным движением или физическими упражнениями, то есть произвольная двигательная активность является провоцирующим фактором развития этих двигательных нарушений (табл. 3) (Темин П.А., Никанорова М.Ю. (ред.), 1999; Panayiotopoulos C.P., 2010; Wolters E., Baumann C. (Eds.), 2014; ILAE, 2019).
| Показатель | Эпилепсия (лобная) | Неэпилептический пароксизмальный кинезиогенный хореоатетоз |
|---|---|---|
| Стереотипность | Да (моторные судороги, преимущественно односторонние) | Комбинация тонических, дистонических и хореоатеоидных феноменов на одной или обеих сторонах |
| Длительность | Обычно 1–3 мин/абсансы до 30 с | ВОт 10–30 с до 3 мин |
| Частота | Разная – от нескольких в год до десятков в сутки | До нескольких десятков в день в большинстве случаев |
| Другие симптомы | Редко | Часто сочетаются с дизартрией, восходящим взглядом и сенсорной аурой |
| Семейный анамнез | Часто | Пациенты могут иметь доброкачественные инфантильные приступы в 3–8 мес., у 8% – эпилептические припадки у родных |
| Ремиссия | Спонтанная ремиссия — очень редко | Спонтанная ремиссия в возрасте 20–30 лет (почти всегда) |
| Возраст дебюта | Часто в детстве и подростковом возрасте | Обычно 5–12 лет |
| Нарушения сознания | Часто | Никогда |
| Амнезия | Часто | Никогда |
| Провоцирующие факторы | Отсутствуют (в некоторых случаях – фотостимуляция, гипервентиляция) | Произвольные движения, физические упражнения |
| ЭЭГ | Эпилептиформные феномены (спайки, полиспайки, спайк-волновые комплексы), после приступа – супрессия активности | Без изменений |
| Ответ на ПЭП | Да | Да |
Считается, что заболевание связано с мутациями в гене PRRT2. Описаны спорадические или семейные случаи с аутосомно-доминантным наследованием. Пароксизмальный кинезиогенный хореоатетоз может сочетаться с эпилепсией при синдроме семейной детской эпилепсии (синдром ICCA). Часть больных имеет в анамнезе доброкачественные инфантильные приступы в возрасте 3-8 мес. и семейный анамнез эпилепсии в 8% случаев.
Заболевание начинается чаще в возрасте 5-16 лет с наступлением спонтанной ремиссии в 20-30 лет почти у всех больных.
Непроизвольные движения совмещают тонические, дистонические и хореоатетоидные моторные феномены с одной стороны или с обеих сторон продолжительностью от 10-30 с до 3 мин. Частота атак очень высока – может возникать несколько десятков приступов в день. Нередко моторные феномены сочетаются с дизартрией, восходящим взглядом и сенсорной аурой. Сознание у больных во время приступа сохранено в полной мере. ЭЭГ регистрируют нормальную биоэлектрическую активность, но эти больные хорошо реагируют на применение ПЭП.
Эпизодическая атаксия 1-го типа. Из разных типов эпизодических атаксий только 1-й тип может вызвать проблемы дифференциальной диагностики с эпилепсией. Заболевание характеризуется короткими эпизодами атаксии, дизартрии и титубации (грубого тремора) головы. Триггером приступа могут быть внезапные движения, эмоции или интеркуррентные заболевания. Такой эпизод атаксии длится от нескольких секунд до минут. Кроме того, у больных между атаками может возникать непрерывная миокимия (часто), лучше заметная под веками при закрытых глазах, или непрерывные движения пальцев с боку на бок, заметные при вытянутых руках.
Частота приступов – ежедневно, от 1–2 до нескольких раз в сутки. Дебютирует заболевание, в среднем, в детском возрасте и продолжается в течение жизни. ЭЭГ дает мало информации для дифференциальной диагностики: у многих больных регистрируют патологические феномены, поскольку эпизодическую атаксию относят к каналопатиям, и в 10% случаев двигательные расстройства у больных сочетаются с фокальными эпилептическими приступами, которые могут трансформироваться в двусторонние тонико-клонические припадки (Imbrici P. et al., 2007; Wolters E., Baumann C. (Eds.), 2014; ILAE, 2019).
Стереотипии (маньеризм) — повторяющиеся движения, позы или выражения, которые могут быть простыми (качания тела, ушибы головой) или сложными (сложные движения пальцами или разгибание/сгибание запястья). Стереотипии могут возникать у здоровых лиц (быть первичными) и быть одним из проявлений других заболеваний, таких как аутизм, заболевания с нарушениями интеллекта и т.д. (вторичные стереотипии) (Panayiotopoulos C.P., 2010; Baizabal-Carvallo J.F. et al., 2019; ILAE, 2019).
Стереотипии можно отличить от эпилептических автоматизмов по характерным движениям. Кроме того, эпилептические автоматизмы развиваются у пациентов на фоне нарушения осознания, нередко сочетаются с другими типами эпилептических приступов – абсансами или фокальными приступами с нарушением осознания. Эпилептические автоматизмы могут также возникать при сохраненном осознании, при очагах в недоминантной височной доле, однако в таких случаях имеются характерные другие признаки поражения височной доли.
Достаточно информативной является ЭЭГ, особенно при проведении мониторинга. Стереотипии не сопровождаются характерными для эпилептических приступов нейрофизиологическими феноменами (Fisher R.S. et al., 2014; Kamble N.L., Pal P.K., 2016; ILAE, 2019).
Неэпилептические двигательные нарушения у новорожденных и младенцев могут неправильно диагностироваться как эпилептические припадки (дрожание, тонические рефлекторные приступы раннего детства, альтернирующая гемиплегия, доброкачественный пароксизмальный тортиколес, ритмичные поведенческие движения, поведенческие расстройства самоудовлетворения, желудочно-эзофагальный рефлюкс у младенцев, доброкачественный неэпилептический миоклонус раннего детства, гиперэкплексия) и составляют тему отдельной публикации.
Таким образом, двигательные нарушения и эпилепсия имеют достаточно много схожих клинических проявлений, что приводит к значительным дифференциально-диагностическим трудностям даже у высококвалифицированных специалистов. Нет абсолютно патогномоничных признаков эпилептических и неэпилептических приступов. Можно выделить только некоторые общие черты неэпилептических пароксизмальных нарушений:
Лечебная тактика у пациентов с эпилептическими приступами, которые должны быть дифференцированы с пароксизмальными двигательными нарушениями, имеет ряд особенностей. Нельзя назначать ПЭП больным с неопределенным диагнозом эпилепсии до окончательного установления диагноза, поскольку эти препараты могут иметь терапевтический эффект и при ряде пароксизмальных двигательных нарушений, но эпилепсия и двигательные нарушения имеют разный прогноз, разные трудовые ограничения и социальные последствия. Всем больным с подозрением на наличие неэпилептических, двигательных пароксизмальных нарушений необходимо проводить ЭЭГ-видеомониторинг, высокопольную (1,5–3Т) магнитно-резонансную томографию с использованием протокола «Эпилепсия», электромиографию (по показаниям), генетическое исследование.
После установления диагноза «Эпилепсия» назначают ПЭП широкого спектра действия, не оказывающие негативного влияния на когнитивные функции и не требующие длительного титрования дозы. В этом случае препаратом выбора может быть леветирацетам (левицитам), который эффективен при большинстве эпилептических приступов, может эффективно применяться во взрослой и детской практике, не вызывает агрегации приступов (в отличие от карбамазепина, барбитуратов и т.п.), эффективен при миоклонических приступах, имеет минимальные побочные эффекты, может быть сразу назначен в терапевтической дозе (для взрослых — 1000 мг/сут). Кроме того, по данным исследований, леветирацетам эффективен при леводопа-индуцированных дискинезиях и целом ряде других двигательных нарушений, а также при сочетании эпилепсии с двигательными нарушениями (Chatterjee A. et al., 2002; Wolz M. et al., 2010; Ebada M.A. et al., 2019).
УКР. МЕД. ЧАСОПИС, 1 (135), Т. 1 – I/II 2020 | www.umj.com.ua
к списку статей Знайти лікаря





